Dr. Reid: Analyzing Genomes

 Robert Reid, Research Assistant Professor of Bioinformatics in the College of Computing and Informatics, uses computational technology and statistics for biological data analysis with expertise in pathway analysis and genome sequence assembly. He has produced genome assemblies in multiple species, including tomato, blueberry, oat, poison dart frog, starfish and sea cucumbers. His past works include developing open-source software visualization tools to create stunning visual linkage map renderings. Utilizing University Research Computing resources, Dr. Reid and his collaborators saved valuable time and money analyzing massive data sets.
Robert Reid, Research Assistant Professor of Bioinformatics in the College of Computing and Informatics, uses computational technology and statistics for biological data analysis with expertise in pathway analysis and genome sequence assembly. He has produced genome assemblies in multiple species, including tomato, blueberry, oat, poison dart frog, starfish and sea cucumbers. His past works include developing open-source software visualization tools to create stunning visual linkage map renderings. Utilizing University Research Computing resources, Dr. Reid and his collaborators saved valuable time and money analyzing massive data sets.
(Pictured: Dr. Robert Reid, Research Assistant Professor of Bioinformatics)
Exploring tissue regeneration using echinoderms
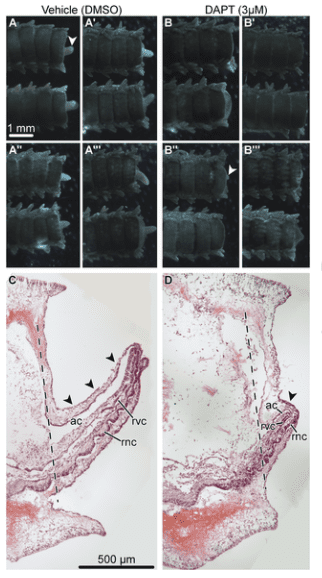 In response to trauma, damaged adult tissue cells have to be activated and instructed to re-enter the cell cycle and eventually differentiate into specialized cell types of a new body part. As regeneration progresses (as seen in the pictured microscopy image of regenerating foot), most of the newly generated cells must stop dividing and migrating to differentiate into specialized cell types. Therefore, regeneration requires complex changes in cell dynamics, which must be tightly coordinated in space and time by genetically encoded signaling pathways. Scientists explore this phenomenon through gene expression sequencing to identify how regeneration occurs.
In response to trauma, damaged adult tissue cells have to be activated and instructed to re-enter the cell cycle and eventually differentiate into specialized cell types of a new body part. As regeneration progresses (as seen in the pictured microscopy image of regenerating foot), most of the newly generated cells must stop dividing and migrating to differentiate into specialized cell types. Therefore, regeneration requires complex changes in cell dynamics, which must be tightly coordinated in space and time by genetically encoded signaling pathways. Scientists explore this phenomenon through gene expression sequencing to identify how regeneration occurs.
“Sea stars and many other echinoderms are highly regenerative species that can regrow almost all tissue types,” Dr. Reid explained. “These features make echinoderms particularly attractive model organisms in regenerative biology.”
As part of the Echinoderm Tree of Life project of the National Science Foundation, collaborators provided data from 43 different species. Dr. Reid, along with project lead Dr. Dan Janies, The Carol Grotnes Belk Distinguished Professor of Bioinformatics and Genomics from CCI, processed, analyzed, and clustered multiple species simultaneously using UNC Charlotte’s High-Performance Computing. The team identified highly expressed genes of interest across species via orthologous similarities in a few short weeks.
“Without the computing capabilities of the HPC, this task would have taken many months,” said Dr. Reid.
To date, the Echinoderm Tree of Life project team has produced 750,000 “orthoclusters” of similar sequences, a computationally demanding step requiring comparisons of over 5 million sequences.
Identifying how tomatoes tolerate heat stress from climate change
 Climate change has placed a challenge on crop species globally. For tomato crops, high temperatures during the pollination stage adversely affect yield productivity. Researchers explore the molecular basis of adaptations during crop reproduction.
Climate change has placed a challenge on crop species globally. For tomato crops, high temperatures during the pollination stage adversely affect yield productivity. Researchers explore the molecular basis of adaptations during crop reproduction.
One National Science Foundation-funded project’s ultimate goal is to develop resources to produce new crop varieties that are viable even at high-temperature stress. These resources 1) detail reproductive gene expression responses to elevated temperature, and 2) register genetic variants across hundreds of tomato genomes to enable analyses of heat stress adaptation and other traits. Sequenced data is aligned to known references, clustered based on similarity, annotated, and counted to identify genic regions that are active under stress conditions.
“Researchers are creating large amounts of genomic sequencing data that we aim to make readily available in community databases,” said Dr. Reid. “A great, concerted computational effort is required to filter, process, and analyze data to produce results.”
Tackling the oat genome with massive sequence data assembly
As population sizes increase, scientists continue to seek how humans can best utilize resources like oats, an excellent dietary fiber source and a breakfast staple in the U.S. A well-established reference genome is the key to speeding up the breeding process and improving breeding efforts, but the oat’s sequence is quite complicated.
“The oat plant genome is a hexaploid that is four times larger than the human genome and consists of six nearly identical copies of highly similar DNA sequences,” said Dr. Reid. “The massiveness of the oat genome makes referencing the genome particularly challenging.”
Assembling the oat genome from small sequence fragments requires a supercomputer with high memory capabilities, large disk arrays for temporary storage, and multiple processor work cycles.
“Previous attempts using popular bioinformatics tools required 40,0000+ job submissions, more than 2TB RAM, and 200TB of temporary data storage using the HPC,” explained Dr. Reid.
Forging ahead
While sequencing technologies improve, and the price of sequencing decreases, the need for adequate computing resources continues to grow. By harnessing the HPC’s power, UNC Charlotte faculty and researchers can work to break through previous computational barriers.